

GSAV: Viele Konsequenzen – auch für die Versorgung
http://doi.org/10.24945/MVF.04.19.1866-0533.2152
>> Die größte Rolle im GSAV spielt, wie aus dem Namen des Gesetzes schon hervorgeht, die Sicherheit der Arzneimittel, eine gesundheitspolitische Konsequenz aus den Skandalen um gefälschte oder verunreinigte Arzneimittel der letzten Jahre. Ebenfalls prominent ist die gesetzlich jetzt festgeschriebene verbindliche Einführung des elektronischen Rezepts. Auch Neuregelungen zum Umgang mit Import-Arzneimitteln, zu neuartigen Therapien und zur Versorgung von Hämophilie-Patienten enthält das neue Gesetz (vgl. Abb. 1). Dadurch betreffen die Maßnahmen zahlreiche an der Arzneimittelversorgung Beteiligte und letztlich auch die Patienten. Denn die Regelungen sollen Arzneimittel nicht nur sicherer machen, sondern auch die Versorgung der Patienten verbessern.
Differenzierte Regelungen bei Importarzneimitteln
Importarzneimittel sind aus dem Ausland importierte Arzneimittel, die aufgrund des Preis-unterschiedes in der Europäischen Union güns-tiger an Apotheken abgegeben werden können als unmittelbar im Inland produzierte (vgl. vdek, 2019). Einen nennenswerten Preisunterschied weisen vor allem patentgeschützte Arzneimittel auf, so dass in diesem Segment die Abgabe eines Importarzneimittels einen Beitrag zur Erfüllung von Wirtschaftlichkeitsreserven leisten kann. So waren Apotheken bisher laut § 129 SGB V Rahmenvertrag zur Arzneimittelversorgung verpflichtet, pro Krankenkasse und Quartal eine Importquote von 5% zu erfüllen. Ein Importarzneimittel musste abgegeben werden, wenn der für die Versicherten maßgebliche Abgabepreis mindestens 15 Prozent oder mindestens 15 Euro niedriger lag als der Preis des Bezugsarzneimittels (vgl. AOK Bundesverband, 2016). Der seit 01. Juli 2019 geltende neue Rahmenvertrag sieht bereits vor Inkrafttreten des GSAV folgende Staffelung bei Arzneimitteln ohne Rabattvertrag vor: Die Abgabe eines preisgünstigen Importarzneimittels ist relevant, wenn bei einem Abgabepreis bis einschließlich 100 Euro ein Preisabstand von mindestens 15%, bei einem Abgabepreis von über 100 Euro bis einschließlich 300 Euro ein Preisabstand von mindestens 15 Euro und bei einem Abgabepreis über 300 Euro ein Preisabstand von mindestens 5% zum Referenzarzneimittel vorliegt. § 13 Absatz 5 des Rahmenvertrags setzt mit diesen Regelungen ein Einsparziel von 2% fest. Importarzneimittel machen im MAT (Moving Annual Total) Mai 2019 mit ca. 20 Mio. Verordnungen einen Anteil von 2,9% aller abgegebenen Packungen am gesamten Fertigarzneimittelmarkt der GKV aus. Damit stehen sie für einen Umsatz von 3,25 Mrd. Euro (nach AVP) im selben Betrachtungszeitraum. Dies entspricht einem Umsatzanteil von 7,8% am Gesamtmarkt (Quelle: NVI, INSIGHT Health). Wurde in den Debatten und den Stellungnahmen zum GSAV noch um die komplette Abschaffung der Importförderklausel gerungen, sieht der jetzige Beschluss differenzierte Regelungen vor und schließt Biopharmazeutika und zytostatische Zubereitungen aus. Zudem hat laut Gesetz der GKV-Spitzenverband dem Bundesministerium für Gesundheit bis Ende 2021 einen Bericht über die Auswirkungen vorzulegen, auf dessen Basis die jetzigen Regelungen überprüft, bewertet und gegebenenfalls über eine Fortführung entschieden werden soll.
Übergangsfrist zur Substitution bei Biosimilars
Ähnlich zur Importförderklausel entspannte sich auch um die automatische Substitution bestimmter Biologika in der Apotheke eine kontroverse Debatte. Dem Vorschlag, auch Biosimilars künftig analog zu den Generika auszutauschen, folgten Änderungsanträge unterschiedlichster Akteure des Gesundheitssystems. In einer Anhörung der Fachverbände hatte lediglich die GKV für die Substitution gestimmt. So verschiebt die geltende Fassung des GSAV den Austausch um drei Jahre. Jedoch soll der G-BA bereits ein Jahr nach Inkrafttreten des Gesetzes in den Richtlinien nach § 92 Absatz 1 Hinweise für die ärztliche Verordnung zur Austauschbarkeit von biologischen Referenzarzneimitteln durch im Wesentlichen gleiche biotechnologisch hergestellte biologische Arzneimittel unter Berücksichtigung ihrer therapeutischen Vergleichbarkeit bestimmen. Spätestens drei Jahre nach Inkrafttreten folgen dann die Hinweise zur Austauschbarkeit von biologischen Referenzarzneimitteln durch Apotheken. Entscheidend für die Definition und damit die Versorgung ist also die Austauschbarkeit in Bezug auf ein bestimmtes Referenzarzneimittel. Damit kann eine automatische Substitution auf adäquate Therapiealternativen eingeschränkt werden. Doch vorerst bleibt mit dieser Übergangsregelung die Therapiehoheit bei den Ärzten.
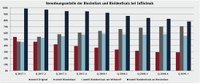
Rein auf die Anzahl der derzeit auf dem deutschen Markt verfügbaren Biosimilars bezogen, betreffen die zukünftigen Regelungen des GSAV nur einen sehr kleinen Teil des Biopharmazeutika-Marktes. Denn bei insgesamt 153 biotechnologisch hergestellten Wirkstoffen (ohne Impfstoffe, Testdiagnostika, Radiopharmazeutika) gibt es lediglich zu 13 Wirkstoffen Biosimilars – derzeit 38 Präparate an der Zahl. Basierend auf Verordnungen in der GKV liegt der Biosimilaranteil am gesamten biosimilarfähigen Markt bei 12,3% (MAT Mai 2019). Der Anteil der untereinander austauschbaren Biosimilars, sprich der Bioidenticals (Definition gemäß Rahmenvertrag nach § 129 Absatz 2 SGB V, Anlage 1), liegt mit 3,7% noch deutlich darunter. Ein Beispiel für die Marktverschiebungen zwischen Original, Biosimilars und Bioidenticals ist in Abbildung 2 für den Wirkstoff Infliximab dargestellt. Der Verordnungsanteil des Originals Remicade fällt erwartungsgemäß stetig, bei zunehmendem Anteil der Biosimilars bis auf 70,8% im 1. Quartal 2019. Gleichzeitig dominieren die beiden Bioidenticals Remsima und Inflectra mit steigenden Anteilen an allen Infliximab-Verordnungen, verlieren aber kontinuierlich in Bezug auf alle Biosimilars. Denn insbesondere das Biosimilar Flixabi weist in den letzten vier Quartalen steigende Verordnungen auf. Und auch das neueste Biosimilar Zessly kommt langsam im Markt an (Quelle: NVI-Plus, INSIGHT Health).
Um besser einschätzen zu können, welche Auswirkungen die automatische Substitution von Biosimilars in der Apotheke haben wird, sind die Bestimmungen des G-BA zur Austauschbarkeit von biologischen Referenzarzneimitteln, auch unter der erwähnten Berücksichtigung ihrer therapeutischen Vergleichbarkeit, abzuwarten. Das Gremium selbst hat in seiner Stellungnahme zum GSAV vom April 2019 die vorgesehene Stärkung des Austausches von biologischen Arzneimitteln begrüßt. Der G-BA formulierte außerdem die Option, zur Umsetzung des Regelungsauftrages und insbesondere zur Beurteilung der therapeutischen Vergleichbarkeit, Auskünfte des BfArM oder des Paul-Ehrlich-Instituts einzuholen. Ob der im Gesetz vorgesehene Zeitraum von ein bzw. drei Jahren für eine Bewertung ausreicht, bleibt also abzuwarten.
Hämophilie-Arzneimittel
Patienten mit Hämophilie haben heute dank guter Therapieoptionen eine Lebenserwartung
nahe dem Bevölkerungsdurchschnitt. In Deutschland sind ca. 5.000 bis 6.000 Patienten von Hä-
mophilie A oder B betroffen. Neben den aus Blutspenden gewonnenen Gerinnungsfaktor-Präparaten stehen auch gentechnisch hergestellte Alternativen zur Verfügung (vgl. PEI 2019).
Mit dem GSAV wird für die Gruppe der gentechnisch hergestellten Arzneimittel die bisherige Ausnahme vom Apothekenvertriebsweg, sprich der Direktvertrieb des Herstellers mit Ärzten und Krankenhäusern, zurückgenommen. Die Prozesse werden damit an die Regelungen der übrigen Biologika angepasst. Sie betreffen aber explizit nicht den Notfallvorrat, den ärztliche Einrichtungen mit einer Spezialisierung auf Hämophilie, in ihren Räumlichkeiten vorhalten und an Patienten oder Einrichtungen der Krankenversorgung abgeben dürfen. Eine Preistransparenz im Hämophilie-Markt will das GSAV durch das Einholen der Herstellerabgabepreise unter Berücksichtigung der abgegebenen Mengen, die sowohl der pharmazeutische Unternehmer als auch die abrechnenden Krankenkassen an den GKV-SV übermittelt, herstellen. Mit diesen Informationen wird nach einer Plausibilitätsprüfung der Herstellerabgabepreis übernommen oder vom GKV-SV festgesetzt.
Durch den Vertriebsweg über die Apotheke entstehen für diese allerdings neue Meldepflichten. Im Falle der Hämophilie-Präparate sind unter anderem die Bezeichnung und weitere Details zum abgegebenen Arzneimittel sowie die Stammdaten des Patienten von der Apotheke an das Deutsche Hämophiliezentrum zu übermitteln. Dies hatte in der Vergangenheit der behandelnde Arzt übernommen. Zum neu festgelegten Vertriebsweg soll die erweiterte Dokumentationspflicht bei allen spezifischen zur Hämophilie-Therapie zugelassenen Arzneimitteln für mehr Sicherheit und Transparenz in der Versorgung sorgen. Damit die langfristige Wirksamkeit und Sicherheit der Therapie besser bewertet werden kann, wird auch die ärztliche Meldepflicht und entsprechend das Deutsche Hämophilieregister beim Paul-Ehrlich-Institut erweitert (vgl. DAZ, 2019).
Ausblick
Im Hinblick auf verträgliche Lösungen war das Ringen der Akteure um ganze Kapitel, einzelne Abschnitte und Details im GSAV bis zu dessen Verabschiedung groß. Im Gesetz festgeschriebene Zeitpunkte bis zur Umsetzung bestimmter Vorgaben und Fristen scheinen im gesamten Prozess Raum für Kompromisse geschaffen zu haben. Das wohl bekannteste Zitat in diesem Zusammenhang „Patienten müssen sich darauf verlassen können, dass Medikamente heilen und ihnen nicht schaden“, stammt von Jens Spahn. Und auch Martin Litsch (Vorstand AOK Bundesverband) befand trotz zahlreicher Kritikpunkte: „Das Gesetz ist der absolut richtige Schritt und enthält viele gute Regelungen, um die Sicherheit von Arzneimitteln zu verbessern.“ Die nächsten Monate werden zeigen, wie die Inhalte in der Praxis umgesetzt werden und welche konkreten Schritte dazu notwendig sind. <<
Autorin: Kathrin Pieloth*
Zitationshinweis:
Pieloth, K.: „GSAV: Viele Konsequenzen – auch für die Versorgung“, in: „Monitor Versorgungsforschung“ (04/19), S. 10-11.; doi: 10.24945/MVF.04.19.1866-0533.2152


